25. Concepts of R2: did you know..?¶
Did you know… that throughout the R2 manual many tips and tricks are provided in small blocksof text containing practical guidance and theoratical background for the analysis at hand? These explanetory blocks start with the phrase “Did you know..”. This chapter aims to centralize the information of the most essential concepts and settings of R2, such that it is easy to integrate these options and understandings in your own analysis.
Jump to one of the sections:
- Statistical terms used in r2 explained?
- Statistical tests in Differential expressed genes?
- Often used settings for analyses
- Core concepts of R2
25.1. R and p-values¶
R: is the correlation coefficient; it ranges from -1 to +1, if R > 0 the value of two variables tends to increase or decrease together. If R < 0 the value of X increases if that of Y decreases, if R~0 there is no relation.Perhaps the best way to interpret the value of R is to square it. This is the fraction of the variance in the two variables that is shared. For example, if R^2 = 0.59 then 59% of the variance in Y can be explained by (or goes along with) variation in X.The p-value for this calculation estimates the probability that this is an observation by pure chance; a p-value of 0.01 you can be 99% sure that this is not the case.
Correlation: What are the p and r-values when the correlation between two genes is calculated. The significance of a correlation is determined by t = R/sqrt((1-r^2)/(n-2)), where R is the correlation value and n is the number of samples. Distribution measure is approximately as t with n-2 degrees of freedom.
25.2. Statistical tests¶
In both types of Differential expressed genes (FindDiff) the two groups and the multiple group variant R2 offers several types of statistical tests as already stated in the differental expressed chapter.
- two group differential expression.
- T-test. For normal distributed and continous data, see ANOVA explanation below.
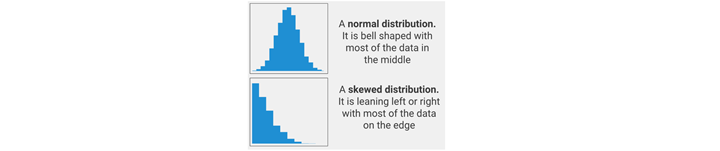
- Mann-Withney-test. The Mann-Whitney U test is used to compare differences between two independent groups when the dependent variable is either ordinal or continuous, but not normally necessarily distributed, such as a skewed distribution.
- Log2 foldchange: How is de log2 foldchange calculated: Log2 (untransformed(group1)/untransformed(grp2))
- Limma: Over the past decade, limma has been a popular choice for gene discovery through differential expression analyses of microarray and high-throughput PCR data, moe information can be found here Limma:NCBI. Limma:BioC.
- DESeq2 algorithm: Differential expression analysis based on the Negative Binomial (a.k.a. Gamma-Poisson) distribution. The algorithm uses raw integer read counts for control and e.g treatment conditions.DESeq2:BioC.
{kind=link}
- Differential expression between groups
- ANOVA: This ANalyis Of VAriance is a statistical test that calculates whether the means of variables differ between two or more groups. In the case of 2 groups, this is identical to the student T-test. ANOVA can be considered a sound test when the variables are normally distributed and samples are independent.
- Kruskal-Wallis: You have non-parametic data and more then 2 groups, in this test the data is replaced by their rank position. I case you want to indentify the difference between specific groups such as group 1 vs 2, 2 vs 3 you should use the Mann-Withney test for two group comparisons.
- Pair-wise tests: T-test is performed for all pair-wise group combinations
25.3. Settings for analyses¶
R2 provides a range of settings for each analysis. Here we explain the background of some recurring concepts and we provide guidance for the use of the different options.
Statistics panel: R2 determines p-values for the differential expression of genes by performing either a one-way anova (default setting) or alternatively a brute-force t-test on any combination of groups when the data is untransformed or log2 transformed. For rank-transformed data, a Kruskal Wallis test is performed. Besides these statistical tests, users can also ask for genes with a certain fold change or obtain a top-X list of the genes which are ordered by a user-specified test.
Correct for multiple testing: Often we are testing a lot of genes in an analysis such as differential expression between groups (Chapter 6). If this is true for your analysis, you have to correct for multiple testing. Why? Let’s look at an example.One might declare that a coin was biased if in 10 flips it landed heads at least 9 times. Indeed, if one assumes as a null hypothesis that the coin is fair, then the probability that a fair coin would come up heads at least 9 out of 10 times is (10 + 1)x(1/2)^ 10 = 0.0107. This is relatively unlikely, and under statistical criteria, such as p-value < 0.05, one would declare that the null hypothesis should be rejected i.e., the coin is unfair.A multiple-comparisons problem arises if one wanted to use this test (which is appropriate for testing the fairness of a single coin), to test the fairness of many coins. Imagine if one was to test 100 fair coins by this method. Given that the probability of a fair coin coming up 9 or 10 heads in 10 flips is 0.0107, one would expect that in flipping 100 fair coins ten times each, to see ;a particular;(i.e., pre-selected) coin come up heads 9 or 10 times would still be very unlikely, but seeing any coin behave that way, without concern for which one, would be more likely than not. Precisely, the likelihood that all 100 fair coins are identified as fair by this criterion is (1 - 0.0107)^100 ~ 0.34.. Therefore the application of our single-test coin-fairness criterion to multiple comparisons would be more likely to falsely identify at least one fair coin as unfair. This occurs in a similar way if we are testing multiple genes in one experiment; we have to correct for this. There are several ways to do so;
- A conservative approach is the Bonferroni correction. The correction is based on the idea that if an experimenter is testing n dependent or independent hypotheses on a set of data, then one way of maintaining the familywise error rate is to test each individual hypothesis at a statistical significance level of 1/n times what it would be if only one hypothesis were tested. So, if it is desired that the significance level for the whole family of tests should be (at most) a, then the Bonferroni correction would be to test each of the individual tests at a significance level of a/n.
- The more sophisticated False Discovery Rate controls the expected proportion of false positives. A FDR threshold is determined from the observed p-value distribution, and hence is adaptive to the amount of signal in your data.
Hugo Once (hugoonce): For most analyses genes should only be reported once in a dataset. R2 uses an algorithm called Hugoonce to choose a single probe-set to represent a gene. For each probe set of a gene, the average expression over all samples with a present call (from the MAS5.0 normalization) is calculated (average present signal APS). The probe set with the highest signal is chosen to represent this gene in the analyzed dataset. For every dataset this procedure is repeated, thereby allowing tissue specific selection for probesets to represent a gene. When no call information is available, the average expression of a probeset is used. In platforms other than Affymetrix, we try to generate a similar score if a notion of being expressed is available. Illumina arrays for example can contain such information. For RNA-seq data, FPKM/RPKM/TPM estimates for a particular gene can be 0. These are also defined in R2 to be Absent or not expressed. The expression calls can also be useful to limit the number of tests that need to be performed, and thereby reduce the multiple testing penalties. Genes that are not considered to be expressed in any of the samples in a cohort can be omitted for a valid reason.
Gene Filters: The gene filters allow you to study a specific subset of genes only. There are several domains you can choose from.
- A specific chromosome can be chosen. Note that when a chromosome is chosen, a specific position range can be defined as well.
- Under GeneCategory some predefined categories can be selected, e.g. some examples are known transcription factors or drugtargets. Here you’ll find the categories you’ve defined yourself also. Kegg pathway selects a set of genes present in the KEGG pathway database .
- Gene Ontology selects a group of genes belonging to a specific Gene Ontology category (www.geneontology.org). Note that if you click a category, further choices deeper down the ontology tree are enabled. Click again on the same dropdown menu to view categories further down the tree.
- Genesets are publicly defined sets or sets you’ve constructed yourself (see for detailed instructions the tutorial “Adapting R2 to your needs”). A convenient search functionality is available to find what you’re looking for. Also in this dropdown feature subsets might be provided once a geneset is selected.Combinations are possible as well; this enables you for example to find the developmental genes on chromosome 1.
Transform: Converting expression levels with the “transform” option can help you to gain additional insight. There are several data transformations available. Note that within R2 most of the data in the resources is stores untransformed, such that you can apply all the transformations within the tools itself.
When to choose which transformation?
- “none”: Raw untransformed expression values, as they are represented in the R2 database.
- “2log”: logarithmic values with base of 2. Every increment constitutes twice the amount.
- “rank”: Data transformation in which numerical or ordinal values are replaced by their rank when the data are sorted by expression. This transformation is useful for non-parametric statistical tests.
- “log2 zscore”: 2log transformed data, centered around the average and expressed as the number of standard deviations from the average. This is the most common option.
- “zscore”: raw intensity values, centered around the average and expressed as the number of standard deviations from the average. This transformation is useful when the intensities in R2 are not raw, but for example logfolds as is often the case for aCGH data.
- “mad/mad2log”: Median absolute deviation (on raw values, or log2 transformed values).
- “center/log2center”: Expression values centered around 0 (on raw values, or log2 transformed values).
- “zcore_group”: Coverts the expression levels from the zscore within a group (track). Applicable when e.g technical variation in expression levels is expected. A possible reason could be when samples from the same dataset originate from different centers.
25.4. Core concepts of R2¶
Tracks: In R2 the samples of a dataset can be annotated with e.g clinical data or molecular biology parameters, each group of annotated data is called a “Track” in R2. These tracks can be used to filter datasets, to compare groups of samples, to color scatter plots of samples with meta information, or to correlate genomics patterns in your data to e.g. different phenotypes or demographic characteristics.Tracks are sample, and therefore, dataset specific. Most datasets contain default tracks when they are uploaded in R2, but every user can define their own custom made tracks for any available dataset, disable/adapt the settings for default tracks or store their analysis outcome as a track to use in further track-related analyses. User defined tracks are privately visible, but can be shared in user defined R2 communities with other R2 users.To play around with the concept, check out the tutorial on the One Gene View analysis, the chapter on Annotation Analyses, or find out how to make your ownn tracks in chapter Adapt R2 to Your Needs.
Categories: Gene sets in R2 are called Categories. These categories enable the user to narrow down an analysis to a specific set of genes that is relevant to their biological question (see Gene Filters in the Settings section above). Users can define custom made Categories or store genes that resulted from an analysis for future use. Follow the tutorial Using Signatures for a better understanding of the possibilities of Categories; check out the section on Gene Categories in Adapting R2 to your needs to learn how to create you own Categories.
Signature Score: Within R2, we can convert the behavior of a list of genes into a signature score that can be calculated for all samples within a particular dataset. This signature score is simply defined as the average zscore of a zscore transformed dataset (the standard way of visualizing a heatmap). We like to think of it as a “meta”-gene: a summary expression value of a group of genes for each sample of a dataset. In R2, such scores are automatically generated when one generates heatmaps via the “view a geneset” function. Because each sample of a dataset will be attributed with a value for the signature score, R2 enables the user to store this signature score as a Track for further analyses with the dataset. For a better understanding of Categories as well as Signature Scores, follow the tutorial Using Signatures.